Looking at SARS-CoV-2 Genome with F#
December 26, 2020 3 Comments
(This post is part of the FSharp Advent Calendar Series. Thanks Sergy Thion for organizing it)
I have just finished up a Cellular and Molecular Biology class at University of Colorado – Boulder (“CU Boulder”, not “UC Boulder” for the unaware) and I was smitten by a couple of the lectures about the COVID virus. Briefly, viruses exist to propagate and since they are an obligate intercellular parasite, they need to find a host cell to encode the host cell’s proteins for that end. That means they enter, hijack some cell proteins, replicate, and then leave to find more cells.
The COVID genome doesn’t really set out to kill humans, that is a side effect of the virus entering the cells (mostly in the lungs) via a common binding site called ACE2. Since the virus takes up the binding site, some regular bodily functions are inhibited so lots of people get sick and some people die. Just like farmers in Brazil view destroying the Amazon rain forest as an unfortunate side effect of them creating a farm to support their families (and then have offspring), the virus doesn’t mean to kill all of these people – it’s just business.

From a computational challenge, the COVID genome is 29,674 base pairs long – which is shockingly small.

You can download the genome from NCBI here. This is a reference genome, which means it is represents several samples of the virus for analysis – it is not one particular sample. The download is in FASTA format – which is a pretty common way to represent the nucleotides of a genome.
Firing up Visual Studio Code, I imported the COVID genome and parsed the file to only have the nucleotides
open System
open System.IO
let path = “/Users/jamesdixon/Downloads/sequence.fasta”
let file = File.ReadAllText(path)
let totalLength = file.Length
let prefixLength = 97
let suffixLength = 2
let stringSequence = file.Substring(prefixLength,totalLength-prefixLength-suffixLength)
I then applied some F# magic to get rid of the magic strings and replace them with a typed representation.
type Base = A | C | G | T
let baseFromString (s:String) =
match s.ToLowerInvariant() with
| “a” -> Some A
| “c” -> Some C
| “t” -> Some T
| “g” -> Some G
| _ -> None
let arraySequence = stringSequence.ToCharArray()
let bases =
arraySequence
|> Seq.map(fun c -> c.ToString())
|> Seq.map(fun s -> baseFromString s)
|> Seq.filter(fun b -> b.IsSome)
|> Seq.map(fun b -> b.Value)
I then went back to the original page and started looking at highlights of this genome. For example, the first marker is this:

UTR stands for “Untranslated Region” – basically ignore the first 265 characters.
The most famous gene in this virus is the spike protein

Where “CDS” stands for coding sequences. Using F#, you can find it as
let spikeLength = abs(21563-25384)
let spike = bases |> Seq.skip 21561 |> Seq.take (spikeLength)
spike
Note that I subtracted two from the CDS value – one because sequences are zero based and one because I am using the “skip” function to go to the first element of the subsequence.
Going back to the first gene listed, you can see in the notes “translated by -1 ribosomal frameshift”
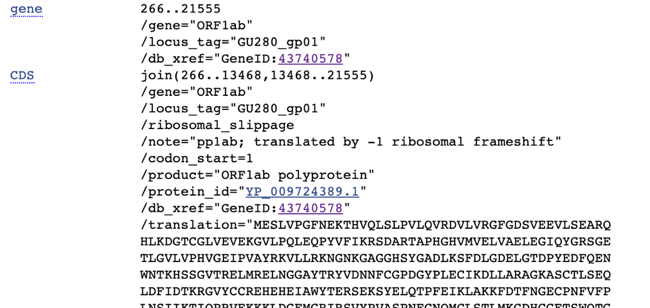
One of the cool things about viruses is that because their genome is so small, they can generate multiple genes out of the same base sequence. They do this with a technique called “frameshifting” where the bases are read in groups of three (a codon) from different start locations – effectively using the same base pair in either the first, second, or their position of a codon. I believe that the ORF1ab gene is read with a frameshift of -1, so the F# would be:
let orf1abLength = abs(266-21555)
let orf1ab = bases |> Seq.skip 263 |> Seq.take (spikeLength)
orf1ab
I subtracted three from the CDS value – one because of zero-based, one for the skip function, and one for the frameshift. I am not 100% that this is the correct interpretation, but it is a good place to start.
I then wanted to look at how the codons mapped to amino acids. I am sure this mapping is done thousands of times in different programs/languages – The F# type system makes the mapping a bit less error prone. Consider the following snippet:
type AminoAcid = Phe | Leu | Iie | Met | Val | Ser | Pro | Thr | Ala | Tyr | His | Gin | Asn | Lys | Asp | Glu
type Codon = Stop | AminoAcid of AminoAcid
I am sure other implementations put the Stop codon with the Amino Acid even though it is not an amino acid. Keeping them separate is more correct – and can prevent bugs later on.
I then started creating a function to map three bases into the correct codon. I initially did something like this:
let AminoAcidFromBases bases =
match bases with
| TTT -> Phe | TTC -> Phe
| TTA -> Leu | TTG -> Leu | CTT -> Leu | CTC -> Leu | CTA -> Leu | CTG -> Leu
| ATT -> Iie | ATC -> Iie | ATA -> Iie
Even though the compiler barfs on this syntax, it is the most intuitive and matches the domain best. I then started coding for the code, rather the domain, by using a sequence like this
let AminoAcidFromBases bases =
match bases with
| [TTT] -> Phe | [TTC] -> Phe
| [TTA] -> Leu | [TTG] -> Leu | [CTT] -> Leu | [CTC] -> Leu | [CTA] -> Leu | [CTG] -> Leu
I also had the problem of incomplete cases – I need the “else” condition like this:
| [] -> None
Which means I then have to go back and put Some in front of all of the results:
| [TTT] -> Some Phe | [TTC] -> Some Phe
But then my code is super cluttered
Also, if I want the function to be “CondonFromBases” versus “AminoAcidFromBasis”, I would have to add this
| [TTT] -> Some (AminoAcid Phe) | [TTC] -> Some (AminoAcid Phe)
And ugh, super-duper clutter.
I am still thinking through the best way to represent this part of the domain. Any suggestions are welcome.
Hopefully this post will inspire some F# people to start looking at computational biology – there are tons of great data out there and lots of good projects with which to get involved.
Gist is here
