When Azure Functions first came out, F# had pretty good support – templates, the ability to run a .fsx file, cool examples written by Don… Fast forward to 2021 and we have bupkis. I recently wrote a dictionary of amino acid weights that would be perfect as a service: pass in a kmer length and weight, get all hits from amino acids.
I first attempted to create a function app, select a .fsx template, and write the code in my browser. Alas, only .csx scripting is available.
I attempted to remove the .csx file and put a .fsx file in via Kudu, but it looks like support for F# scripting is not installed. I created an issue on Azure User voice, but Jordan Marr beat me to it by about 2 years.
I then attempted to open VS Code and upload a function to Azure. Alas, only C# templates are available in VS Code and replacing .fs and .fsproj files in the OOB project fails on compile.
I then installed Visual Studio (remember that?) hoping that it might have some templates. Nope.
So I ranted on Twitter and I got some helpful advice from Jon Canning and Jiri Landsman: both have projects on Git Hub that create an Azure Function: here and here. I downloaded them – Jon’s took some work because I didn’t have Paket installed. I am amazed that for a simple “Hello World” return from a http request takes 11 files and of the two files that have F# code, there are 31 lines of code. That is about 1 file and two lines of code per character.
I got both project building locally, but I was at a loss about how to actually deploy them to Azure since there was not a built-in facility in VS Code or Visual Studio. Jon pointed me to this article where I figured out the deployment.
Here are the steps to get a F# Azure Function working:
Go into Azure and create a Azure Function App
Download either Jon or Jiri’s template
Build the template locally
Go to the bin folder and compress the contents. Do not compress the folder itself.
Drag/Drop the .zip file you made in #4 into the screen. Wait for Kudu to finish
Go to your function app and hopefully see your new function
Navigate to that function to see it working
Lament the fact that you have to jump though all of these hoops when all you wanted to do was just return “hello world” to a http request
I have not tried deploying to Azure from GitHub yet. If past experience is any indication, I will need to carve out a Saturday to do it. What really annoys me is that I have a limited time to work on this project and instead of actually writing code that will help cure diabetes, I am spending it doing this garbage. No wonder academics just do everything on their machine. Hopefully Microsoft will recognize this fact and devote more help to people trying to solve domain problems so we can spend less time worrying about solving Microsoft Infrastructure puzzles.
The heart of hypedsearch is a “seed and extend” algorithm. We take the results from the mass spectrometry and compare them to our known dictionary of peptides to see if any are hybrid. The problem is that the mass-spec observations can be 1 to n amino acids and there is considerable noise in the samples. As a way of increasing compute time performance, we decided to break the dictionary of known peptides into a catalog of fragments with each catalog based on the size of the kmer used to break the protein’s amino acid chains into parts. The idea is to pass a weight from a mass-spec observation and get a return of all possible peptides that have the same weight (measured in daltons)
I first implemented the dictionary is python since that is what hypedsearch is written in. The proteins are stored in a file format called “.fasta” which is the de-facto standard for storing genomic and proteomic data. There is actually a fasta module that makes reading and parsing the file a snap. After reading the file, I parsed the contents into a data structure that contains the name of the protein and a collection of amino acids – each with their individual weight
The next step is to create a data structure that has a attribute of weight and the amino acids associated with that weight – with the index from the original protein of where that amino acid chain is located (note that I used amino acid chain and peptide interchangeably, apologies if some biologist out there just threw their Cheetos at the screen).
Once I had all of the proteinweights for a given kmer, I could then bundle them up into a data structure that had all of the records associated with a single weight.
Weight_Protein_Matches = namedtuple('Weight_Protein_Matches',['Weight','Protein_Match'],defaults=[0,[]])
def handle_weight_protein_matches(weight_protein_matches, all_weight_protein_matches):
exists = False
for item in all_weight_protein_matches:
if item.Weight == weight_protein_matches.Weight:
item.Protein_Match.append(weight_protein_matches)
exists = True
break
if exists == False:
all_weight_protein_matches.append(weight_protein_matches)
def generate_all_weight_protein_matches(protein_matches):
all_weight_protein_matches = []
for protein_match in protein_matches:
weight = protein_match.Weight
weight_protein_matches = Weight_Protein_Matches(weight, [protein_match])
handle_weight_protein_matches(weight_protein_matches,all_weight_protein_matches)
return all_weight_protein_matches
Nothing really exciting here (the way code should be) – just lots of loops. I did try to avoid mutable variables and I am not happy with that one early return in handle_weight_protein_matches. I then took the functions out for a spin.
file_path = '/Users/jamesdixon/Documents/Hypedsearch_All/hypedsearch/data/database/sample_database.fasta'
fasta_parser = fasta.read(file_path) #n = 279
proteins = generate_proteins(fasta_parser)
proteins_matches = generate_proteins_matches(proteins,2) #n=103163 for kmer=2
all_weight_protein_matches = generate_all_weight_protein_matches(proteins_matches) #n=176 for kmer=2 and round to 2 decimal places
print(all_weight_protein_matches)
And it ran like a champ
So then I thought “I am not a fan of all of those loops and named tuples for data structures leaves a lot to be desired. I wonder if I can implement this in F#? Also I was inspired by Don teaching Guido F# last week might have entered my thinking. Turns out, it is super easy in F# thanks to the high-order functions in the language.
Step one was to read the data from the file. Unlike python, there is a not a .fasta type provider AFAIK so I wrote some code to parse the contents (thank you to Fyodor Soikin for answering my Stack Overflow question on the chunk function)
type FastaEntry = {Description:String; Sequence:String}
let chunk lines =
let step (blocks, currentBlock) s =
match s with
| "" -> (List.rev currentBlock :: blocks), []
| _ -> blocks, s :: currentBlock
let (blocks, lastBlock) = Array.fold step ([], []) lines
List.rev (lastBlock :: blocks)
let generateFastaEntry (chunk:String List) =
match chunk |> Seq.length with
| 0 -> None
| _ ->
let description = chunk |> Seq.head
let sequence = chunk |> Seq.tail |> Seq.reduce (fun acc x -> acc + x)
Some {Description=description; Sequence=sequence}
let parseFastaFileContents fileContents =
chunk fileContents
|> Seq.map(fun c -> generateFastaEntry c)
|> Seq.filter(fun fe -> fe.IsSome)
|> Seq.map(fun fe -> fe.Value)
Generating the amino acids and proteins was roughly equivalent to the python code – though I have to admit that the extra characters when setting up the amino acid record types was annoying compared to the name tuple syntax. On the flip side, no for..each – just high order .skip, .head, .map to do the work and .toList to keep the data aligned.
type ProteinMatch = {ProteinName:string; Weight:float; StartIndex:int; EndIndex:int}
let generateProteinMatch (protein: Protein) (index:int) (aminoAcids:AminoAcid array) (kmerLength:int) =
let name = protein.Name
let weight =
aminoAcids
|> Array.map(fun aa -> aa.Weight)
|> Array.reduce(fun acc x -> acc + x)
let startIndex = index * kmerLength
let endIndex = index * kmerLength + kmerLength
{ProteinName = name; Weight = weight; StartIndex = startIndex; EndIndex = endIndex}
let generateProteinMatches (protein: Protein) (kmerLength:int) =
protein.AminoAcids
|> Seq.windowed(kmerLength)
|> Seq.mapi(fun idx aa -> generateProteinMatch protein idx aa kmerLength)
let generateAllProteinMatches (proteins: Protein list) (kmerLength:int) =
proteins
|> Seq.map(fun p -> generateProteinMatches p kmerLength)
|> Seq.reduce(fun acc pm -> Seq.append acc pm)
So I love the windowed function for creating the slices of kmers. Compared to the python, I find the code is much more readable and much more testable – plus the bonus of parallelism by adding “PSeq”.
More love: the groupBy function for the summation is perfect. The groupBy function in python does not return a similar data structure of the index and the associated collections – the way F# does it makes building up that data structure a snap. Less Code, fewer errors, more fun.
Once the functions were in place, I took them out for a spin
let filePath = @"/Users/jamesdixon/Documents/Hypedsearch_All/hypedsearch/data/database/sample_database.fasta"
let fileContents = File.ReadLines(filePath) |> Seq.cast |> Seq.toArray
let parsedFileContents = parseFastaFileContents fileContents
let proteins = generateProteins parsedFileContents
let proteinMatches = generateAllProteinMatches proteins 2
let weightProteinMatches = generateWeightProteinMatches (proteinMatches |> Seq.toList)
And it ran like a champ
Since I am obviously a F# fan-boy, I much preferred writing the F#. I am curious what my python-centric colleges will think when I present this…
When I first started getting interested in Computational Biology, I had a very simplistic mental model of genes based on the “central dogma of biology”: DNA makes RNA and RNA makes proteins. I thought that the human genome was just computer code – instead of binary ones and zeros, it was base-four “A” “T” “C” “G”.*. Therefore, to understand it, just start mapping the genome like the human genome project did and then see the areas that I am interested in: the four billion nucleotides are memory on the hard drive with some of the genome being the operating system and some being installed software. When the computer is powered on, the ones and zeros are read off of the hard drive and placed into memory – the equivalent would be the DNA being read out of the chromosomes and placed into RNA.
Man, was I wrong.
DNA is not just your operating system and installed programs – it is the complete instructions to make the computer, turn it on, write, compile, and then run the most sophisticated software in the world. Oh, it also has the instructions to replicate itself and correct errors that might be introduced during replication. And we can barely figure out how to write Objective C for a phone app…
So carrying the analogy of the human genome to computer, even if we could determine which part of bytes on the hard drive is for building a transistor, for example, we have another problem. Unlike a computer’s hard drive where each byte is allocated to only one purpose, each nucleotide can be used by none, one, or many genes. It is a fricken madhouse – a single “C-G” pair might be used in creating a val amino acid in one protein for one gene, frameshifted to creating an ala amino acid in a different protein for another gene, and then be used in the regulation region of yet another gene being read in the reverse direction.
The implication is that it does not appear possible to “build up” and domain model of the genome. You can’t take a series of nucleotides of DNA like “AAAGGTTCGTAGATGCTAG” and know anything about what it is doing. Rather, it looks like the domain model has to work in reverse: Given a gene like TP53, allocate different sections of DNA to the different regions: Promoter, Intons, Exons, UTRs, etc….
From a F# modeling point of view, DNA is certainly an “OR” type:
type DNA = A | T | C| G
With the caveat that we sometimes don’t know if a location is really an A, rather it is not a G or T. Sweeping aside that problem, then individual nucleotides are an “AND” type:
Type Nucleotide = {Index: int; DNA: DNA; etc…}
let nucleotide0 = {Index=0; DNA=C; etc…}
and then gene would look something like this:
let tP53 = {Gene= TP53; Promoter = [nucleotide0; nucleotide1]; etc…}
Note that I am only 1/3 of the way through my genetics class right now, so I might change my understanding next week. For now, this is my Mental Model 0.2 of the genome.
*side note: some April 4th, I want to sneak into all of the computational biologists offices and steal the A,C,T,G,Us keys from their keyboards. Pandemonium would reign.
(This post is part of the FSharp Advent Calendar Series. Thanks Sergy Thion for organizing it)
I have just finished up a Cellular and Molecular Biology class at University of Colorado – Boulder (“CU Boulder”, not “UC Boulder” for the unaware) and I was smitten by a couple of the lectures about the COVID virus. Briefly, viruses exist to propagate and since they are an obligate intercellular parasite, they need to find a host cell to encode the host cell’s proteins for that end. That means they enter, hijack some cell proteins, replicate, and then leave to find more cells.
The COVID genome doesn’t really set out to kill humans, that is a side effect of the virus entering the cells (mostly in the lungs) via a common binding site called ACE2. Since the virus takes up the binding site, some regular bodily functions are inhibited so lots of people get sick and some people die. Just like farmers in Brazil view destroying the Amazon rain forest as an unfortunate side effect of them creating a farm to support their families (and then have offspring), the virus doesn’t mean to kill all of these people – it’s just business.
From a computational challenge, the COVID genome is 29,674 base pairs long – which is shockingly small.
You can download the genome from NCBI here. This is a reference genome, which means it is represents several samples of the virus for analysis – it is not one particular sample. The download is in FASTA format – which is a pretty common way to represent the nucleotides of a genome.
Firing up Visual Studio Code, I imported the COVID genome and parsed the file to only have the nucleotides
open System
open System.IO
let path = “/Users/jamesdixon/Downloads/sequence.fasta”
let file = File.ReadAllText(path)
let totalLength = file.Length
let prefixLength = 97
let suffixLength = 2
let stringSequence = file.Substring(prefixLength,totalLength-prefixLength-suffixLength)
I then applied some F# magic to get rid of the magic strings and replace them with a typed representation.
type Base = A | C | G | T
let baseFromString (s:String) =
match s.ToLowerInvariant() with
| “a” -> Some A
| “c” -> Some C
| “t” -> Some T
| “g” -> Some G
| _ -> None
let arraySequence = stringSequence.ToCharArray()
let bases =
arraySequence
|> Seq.map(fun c -> c.ToString())
|> Seq.map(fun s -> baseFromString s)
|> Seq.filter(fun b -> b.IsSome)
|> Seq.map(fun b -> b.Value)
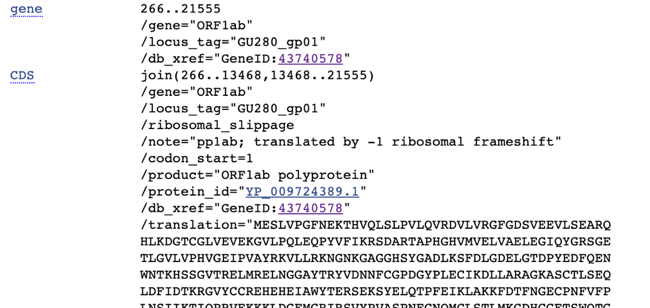
I then went back to the original page and started looking at highlights of this genome. For example, the first marker is this:
UTR stands for “Untranslated Region” – basically ignore the first 265 characters.
The most famous gene in this virus is the spike protein
Where “CDS” stands for coding sequences. Using F#, you can find it as
let spikeLength = abs(21563-25384)
let spike = bases |> Seq.skip 21561 |> Seq.take (spikeLength)
spike
Note that I subtracted two from the CDS value – one because sequences are zero based and one because I am using the “skip” function to go to the first element of the subsequence.
Going back to the first gene listed, you can see in the notes “translated by -1 ribosomal frameshift”
One of the cool things about viruses is that because their genome is so small, they can generate multiple genes out of the same base sequence. They do this with a technique called “frameshifting” where the bases are read in groups of three (a codon) from different start locations – effectively using the same base pair in either the first, second, or their position of a codon. I believe that the ORF1ab gene is read with a frameshift of -1, so the F# would be:
let orf1abLength = abs(266-21555)
let orf1ab = bases |> Seq.skip 263 |> Seq.take (spikeLength)
orf1ab
I subtracted three from the CDS value – one because of zero-based, one for the skip function, and one for the frameshift. I am not 100% that this is the correct interpretation, but it is a good place to start.
I then wanted to look at how the codons mapped to amino acids. I am sure this mapping is done thousands of times in different programs/languages – The F# type system makes the mapping a bit less error prone. Consider the following snippet:
type AminoAcid = Phe | Leu | Iie | Met | Val | Ser | Pro | Thr | Ala | Tyr | His | Gin | Asn | Lys | Asp | Glu
type Codon = Stop | AminoAcid of AminoAcid
I am sure other implementations put the Stop codon with the Amino Acid even though it is not an amino acid. Keeping them separate is more correct – and can prevent bugs later on.
I then started creating a function to map three bases into the correct codon. I initially did something like this:
let AminoAcidFromBases bases =
match bases with
| TTT -> Phe | TTC -> Phe
| TTA -> Leu | TTG -> Leu | CTT -> Leu | CTC -> Leu | CTA -> Leu | CTG -> Leu
| ATT -> Iie | ATC -> Iie | ATA -> Iie
Even though the compiler barfs on this syntax, it is the most intuitive and matches the domain best. I then started coding for the code, rather the domain, by using a sequence like this
let AminoAcidFromBases bases =
match bases with
| [TTT] -> Phe | [TTC] -> Phe
| [TTA] -> Leu | [TTG] -> Leu | [CTT] -> Leu | [CTC] -> Leu | [CTA] -> Leu | [CTG] -> Leu
I also had the problem of incomplete cases – I need the “else” condition like this:
| [] -> None
Which means I then have to go back and put Some in front of all of the results:
| [TTT] -> Some Phe | [TTC] -> Some Phe
But then my code is super cluttered
Also, if I want the function to be “CondonFromBases” versus “AminoAcidFromBasis”, I would have to add this
| [TTT] -> Some (AminoAcid Phe) | [TTC] -> Some (AminoAcid Phe)
And ugh, super-duper clutter.
I am still thinking through the best way to represent this part of the domain. Any suggestions are welcome.
Hopefully this post will inspire some F# people to start looking at computational biology – there are tons of great data out there and lots of good projects with which to get involved.
Pressing on with more bioinformatic algorithms implemented in a functional style, the next algorithm found in Bioinformatics Algorithms by Compeau and Pevzner is to find the most frequent pattern in a string of text.
I started writing in the imperative style from the book like so (the length of the substring to be found is called the “k-mer” so it gets the parameter name “k”)
type Count = {Text:string; PatternCount:int}
let frequentWords (text:string) (k:int) =
let patternCount (text:string) (pattern:string) =
text
|> Seq.windowed pattern.Length
|> Seq.map(fun c -> new string(c))
|> Seq.filter(fun s -> s = pattern)
|> Seq.length
let counts = new List<Count>()
for i = 0 to text.Length-k do
let pattern = text.Substring(i,k)
let patternCount = patternCount text pattern
let count = {Text=text; PatternCount=patternCount}
counts.add(count)
counts |> Seq.orderByDesc(fun c -> c.PatternCount)
let maxCount = counts|> Seq.head
let frequentPatterns = new List<Count>()
for i = 0 to counts.length
if count.[i].PatternCount = maxCount then
frequentPatterns.add(count.[i])
else
()
But I gave up because, well, the code is ridiculous. I went back to the original pattern count algorithms written in F# and then added in a block to find the most frequent patterns:
let frequentWords (text:string) (k:int) =
let patternCounts =
text
|> Seq.windowed k
|> Seq.map(fun c -> new string(c))
|> Seq.countBy(fun s -> s)
|> Seq.sortByDescending(fun (s,c) -> c)
let maxCount = patternCounts |> Seq.head |> snd
patternCounts
|> Seq.filter(fun (s,c) -> c = maxCount)
|> Seq.map(fun (s,c) -> s)
The VS Code linter was not happy with my Seq.countBy implementation… but it works. I think the code is explanatory:
window the string for the length of k
2. do a countyBy on the resulting substrings
3. sort it by descending, find the top substring amount
4. filter the substring list by that top substring count.
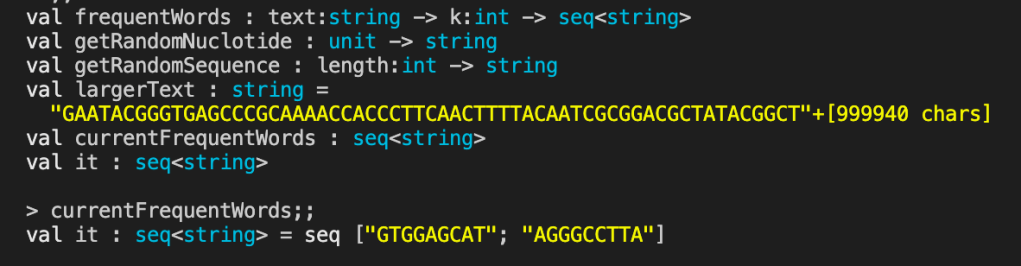
The last map returns just the pattern and leaves off the frequency, which I think is a mistake but is how the book implements it. Here is an example of the frequentWords function in action:
let getRandomNuclotide () =
let dictionary = ["A";"C";"G";"T"]
let random = new Random()
dictionary.[random.Next(4)]
let getRandomSequence (length:int) =
let nuclotides = [ for i in 0 .. length -> getRandomNuclotide() ]
String.Join("", nuclotides)
let largerText = getRandomSequence 1000000
let currentFrequentWords = frequentWords largerText 9
currentFrequentWords
I didn’t set the seed value for generating the largerText string so the results will be different each time.
I noticed in both cases that the code samples are very much imperative focused: lots of loops, lots of mutable variables, lots of mess. I endeavored to re-write the code in a more functional style using immutable variables and pipelining functions
Consider the code for Pattern Count, a function that counts how often a pattern appears in a larger string. For example, the pattern “AAA” appears in the larger string “AAATTTAAA” twice. If the larger string was “AAAA”, the pattern “AAA” is also twice since AAA appears in index 0-2 and index 1-3.
Here is the code that appears in the book:
let patternCount (text:string) (pattern:string) =
let mutable count = 0
for i = 0 to (text.Length - pattern.Length) do
let subString = text.Substring(i,pattern.Length)
if subString = pattern then
count <- count + 1
else
()
count
Contrast that to a more functional style:
let patternCount (text:string) (pattern:string) =
text
|> Seq.windowed pattern.Length
|> Seq.map(fun c -> new string(c))
|> Seq.filter(fun s -> s = pattern)
|> Seq.length
There are three main benefits of the functional style:
The code is much more readable. Each step of the transformation is explicit as a single line of the pipeline. There is almost a one to one match between the text from the book and the code written. The only exception is the Seq.map because the windowed function outputs as a char array and we need to transform it back into a string.
The code is auditable. The pipeline can be stopped at each step and output reviewed for correctness.
The code is reproducible. Because each of the steps uses immutable values, pattern count will produce the same result for a given input regardless of processor, OS, or other external factors.
In practice, the pattern count can be used like this:
let text = "ACAACTCTGCATACTATCGGGAACTATCCT"
let pattern = "ACTAT"
let counts = patternCount text pattern
val counts : int = 2
In terms of performance, I added in a function to make a sequence of ten million nucleotides and then searched it:
let getRandomNuclotide () =
let dictionary = ["A";"C";"G";"T"]
let random = new Random()
dictionary.[random.Next(4)]
let getRandomSequence (length:int) =
let nuclotides = [ for i in 0 .. length -> getRandomNuclotide() ]
String.Join("", nuclotides)
let largerText = getRandomSequence 10000000
#time
let counts = patternCount largerText pattern
Real: 00:00:00.814, CPU: 00:00:00.866, GC gen0: 173, gen1: 1, gen2: 0 val counts : int = 9816
It ran in about one second on my macbook using only 1 processor. If I wanted to make it faster and run this for the four billion nucleotides found in human DNA, I would use the Parallel Seq library, which is a single letter change to the code. That would be a post for another time…
(This post is part of the FSharp Advent Calendar Series. Thanks Sergy Thion for organizing it)
Recently, I had the need to get articles from some United States government websites. You would think in 2019 that these sites might have apis and you would think wrong. In each case, I needed to crawl the site’s HTML and then extract the information. I started doing this with Python and its beautiful soup library but I can into the fundamental problem that getting the html was much harder than parsing the site. To illustrate, consider this website
I need to go through all 8 pages of the grid and download the .pdfs that are associated with the “View Report” link. The challenge in this particular site is that they didn’t do any url parameters so there is no way to go through the grid via the uri. Looking at the page source, they are using ASP.NET and in typical enterprise-derpy manner, named their table “GridView1”
The way to get to the next page is to press on the “Next” link defined like this:
They over-achieved in the bloated View State for a simple page category though.
#Sigh
So as bad as this site is, F# made getting the data a snap. I fired up Visual Studio and created a new .NET Core F# project. I added a script file. I realized that the button-press to get to the next page was going to be a pain to program, so I decided to use the .NET framework WebBrowser class. It’s nice because it has all of the apis I needed for the traversal and I didn’t have to make the control visible.
My first function was to get the uris from the grid – easy enough using the HtmlDocument and HtmlElement classes:
let getPdfUris (document:HtmlDocument) =
let collection = document.GetElementsByTagName(“a”)
collection
|> Seq.cast
|> Seq.filter(fun e -> e.OuterText = “View Report”)
|> Seq.map(fun e -> e.GetAttribute(“href”))
Note the key word I used to filter was “View Report”, so at least the web designer stayed consistent there.
Next, I used basically the same logic to find the Next button in the DOM. Note that I am using the TryFind function so if the button is not found, a None is returned:
let getNextButton (document:HtmlDocument) =
let collection = document.GetElementsByTagName(“a”)
collection
|> Seq.cast
|> Seq.tryFind(fun e -> e.InnerText = “Next”)
So the next function was my giggle moment for this project. To “press” that button to invoke the javascript to go to the next page of the grid, I ised the InvokeMember method of the HtmlClass
let invokeNextButton (nextButton: HtmlElement) =
nextButton.InvokeMember(“Click”) |> ignore
printfn “Next Button Invoked”
Yup, that works! I was worried that I was going to have to screw around with the javascript or, worse, that beast called View State. Fortunately, that InvokeMember method worked fine. Another reason why I love the .NET framework.
So with these three functions set up, I created a method to be called each time the document is refreshed
let handlePage (browser:WebBrowser) (totalUris:List) =
let document = browser.Document
let uris = getPdfUris document
totalUris.AddRange(uris)
let nextButton = getNextButton document
match nextButton with
| Some b ->
invokeNextButton b
| None -> ()
My C#-only friends spend waaaay to much time worrying about the last page and having no button and how to code it. I used the option type – is there a Next button? Press it and do work. No Button? Do nothing.
I put in a function to save the .pdf to my local file system
let downloadPdf (uri:string) =
let client = new WebClient();
let targetFilePath = @”C:\Temp\” + Guid.NewGuid().ToString() + “.pdf”;
So I new up the browser, send handlePage to the DocumentCompleted event handler. Every time the Next button is pressed, the document loads and the DocumentCompleted event fires, and the .pdfs are downloaded and the next button is pressed. Until the last page, when there is no button to press.
As a follow up to this post, I decided to look on the other side of the gun –> police officers killed in the line of duty. Fortunately, the FBI collects this data here. It looks like the FBI is a bit behind on their summary reports:
So taking the 2013 data as the closest data point to The Counted 2015 data, it took a couple of minutes to download the excel spreadsheet and format it as a useable .csv:
to
After importing in the data in R studio, I did a quick summary on the data frame. The most striking thing out of the gate is how few Officers are killed. There were 27 in 2013, compared to over 500 people killed by police officers in the 1st half of 2015:
So Louisiana and West Virginia seem to have the highest number of officers killed per capita. I am not surprised, being that I had no expectations about states that would have higher and lower numbers. It seems likely a case of “gee-wiz” data.
Since there is so few instances, I decided to forgo any more analysis on police killed and instead combined this data with the people who were killed by police:
and certainly the log helps out and there seems to be a relationship between states that have police killed and people being killed by police (my hand-drawn red lines added):
With that in mind, I created a couple of linear models
Since there are only 2 variables, the adjusted R square is the same for x~y and y~x.
The interesting thing is the model has to account that many states had 0 police fatalities but had at least 1 person killed by the police. The next interesting thing is the value of the coefficient: in starts where there was at least 1 police fatality and 1 person killed by the police, every police fatality increases the number of people killed by police .96 –> and this .96 is the log of the ratio of population. So it shows that the police are better at killing then getting killed, which makes sense.
Disclaimer: I really don’t know what I am talking about
I received an email from a coworker/friend yesterday with this in the body:
So, I have a friend who works for a major supermarket chain. In IT, they are straight out of the year 2000. They have tons and tons of data in SQL Server and I think Oracle. The industrial engineers (who do all of the planning) ask the IT group to run queries throughout the day, which takes hours to run. They use Excel for most of their processing. On the weekends, they run reporting queries which take hours and hours to run – all to get just basic information.
This got my wheels spinning about how I would approach the problem with the analytics toolset that I know is available. The supermarket chain has a couple of problems
Lots of data that takes too long to munge through
The planners are dependent on IT group for processing the data
I would expect the official Microsoft answer is that they should implement Sql Server Analytics with Power BI. I would assume if the group threw enough resources at this solution, it would work. I then thought of a couple of alternative paths:
The first thing that comes to mind is using HDInsight (Microsoft’s Hadoop product) on Azure. That way the queries can run in a distributed manner and they can provision machines as they need them -> and when they are not running their queries, they can de-allocate the machines.
The second thought is using AzureML to do their model generation. However, depending on the size of the datasets, AzureML may not be able to scale. I have only used Azure ML on smaller datasets.
The third thought was using R? I don’t think R is the best answer here. Everything I know about R is that it is designed for data exploration and analysis of datasets that comfortably fit into the local machine’s memory. Performance on R is horrible and scaling R is a real challenge.
What about F#? So this might be a good answer. If you use the Hive Type Provider, you can get the benefits of HDInsight to do the processing and then have the goodness of the language syntax and REPL for data exploration. Also, the group could look at MBrace for some kick-butt distributed processing that can scale on Azure. Finally, if they don come up with some kind of insight that lends itself for building analytics or models into an app, you can take the code out of the script file and stick it into a compliable assembly all within Visual Studio.
What about Python? No idea, I don’t enough about it
What about Matlab, SAS, etc.. No idea. I stopped using those tools when R showed up.
What about Watson? No idea. I think I will have a better idea once I go to this.